In the past few decades, pulsed laser sources have become more and more often found in condensed matter physics and physical chemistry labs. A typical use of these sources is a so-called “pump-probe” experiment. In these investigations, an initial laser “pump” pulse excites a sample and a subsequent “probe” pulse then monitors how the sample relaxes back to equilibrium. By varying the time delay between the initial “pump” pulse and the subsequent “probe” pulse, the relaxation can be tracked as a function of time. One of the first remarkable observations seen with this technique is what I show in the figure below — oscillations of a solid’s reflectivity after short-pulse photoexcitation (although many other experimental observables can also exhibit these kinds of oscillations). These oscillations arise from the excitation of a vibrational lattice mode (i.e. a phonon).

Why is this observation interesting? After all, isn’t this just the excitation of a vibrational mode which can be also be excited thermally or in a scattering experiment? In some sense yes, but what makes this different is that the excited phonon is coherent. This means, unlike in the context of a scattering experiment, the atomic motion is phase-locked across the entire photo-excited area; the atoms move back and forth in perfect synchrony. This is why the oscillations show up in the measured observables — the macroscopic lattice is literally wobbling with the symmetry and frequency of the normal mode. (In a scattering experiment, by contrast, the incident particles, be they electrons, neutrons or photons, are continuously shone onto the sample. Therefore, the normal modes are excited, but at different times. These different excitation times result in varying phases of the normal mode oscillations, and the coherent oscillations thus wash out on average.)
There are many different ideas on how to use these coherent oscillations to probe various properties of solids and for more exotic purposes. For example, in this paper, the Shen group from Stanford showed that by tracking the oscillations in an X-ray diffraction peak (from which the length scale of atomic movements can be obtained) and the same vibrational mode oscillations in a photoemission spectrum (from which the change in energy of a certain band can be obtained), one can get a good estimate of the electron-phonon coupling strength (at least for a particular normal mode and band). In this paper from the Ropers group at Gottingen, on the other hand, an initial pulse is used to melt an ordered state through the large amplitude excitation of a coherent mode. A subsequent pulse then excites the same mode out-of-phase leading to a “revival” of the ordered state.
When oscillations of these optical phonon modes first started appearing in the literature in the late 1980s and early 90s, there was a lot of debate about how they were generated. The first clue was that only Raman-active modes showed up; infrared-active oscillations could not be observed (in materials with inversion symmetry). While the subsequent proposed Raman-based mechanisms could explain almost all observations, there were certain modes, like the one in Bismuth depicted in the figure above, that did not conform to the Raman-type excitation scheme. A new theory was put forward suggesting that this mode (and other similar ones) were excited through a so-called “displacive” mechanism.
One distinction between the two generation mechanisms is that the Raman-type theory predicted a sine-like oscillation, whereas the displacive-type theory predicted a cosine-like oscillation (i.e. there was a distinction in terms of the phase). Another prediction of the “displacive” theory was that only totally symmetric modes could be excited in this way (see image below). In the image depicting the oscillations in Bismuth above, an arrow in the inset points to an energy where a vibrational excitation is seen with spontaneous Raman spectroscopy but is not present in the pump-probe experiment. The only visible vibrational mode is the totally symmetric one, consistent with the displacive excitation theory.

In this post, I’m going to go through some toy model-like ideas behind the two generation mechanisms and briefly go over the symmetry arguments that allow their excitation. In particular, I’ll explain the difference between the sine and cosine-like oscillations and also why the “displacive” mechanism can only excite totally symmetric modes.
Impulsive stimulated Raman scattering (ISRS) is the rather intimidating name given to the Raman-type generation mechanism. Let’s just briefly try to understand what the words mean. Impulsive refers to the width of the light pulse, 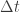
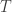


Now that we have a picture of how this process works, let us return to our first question: why does the Raman-type generation process result in a sine-like time dependence? Consider the following equation of motion, which describes a damped harmonic oscillator subject to an external force that is applied over an extremely short timescale (i.e. a delta function):

where 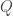
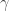


Below is a qualitative schematic of this function:

Seeing the solution to this equation should demonstrate why a short pulse perturbation would give rise to a sine-like oscillation.
So the question then becomes: how can you get something other than a sine-like function upon short-pulse photoexcitation? There are a couple of ways, but this is where the displacive theory comes in. The displacive excitation of coherent phonons (DECP) mechanism (another intimidating mouthful of a term) requires absorption of the photons from the laser pulse, in contrast to the Raman-based mechanism which does not. Said another way, one can observe coherent phonons in a transparent crystal with a visible light laser pulse only through the Raman-based mechanism; the displacive excitation of coherent phonons is not observed in that case.
What this tells us is that the displacive mechanism depends on the excitation of electrons to higher energy levels, i.e. the redistribution of the electronic density after photoexcitation. Because electrons are so much lighter than the nuclei, the electrons can come to equilibrium among themselves long before the nuclei can react. As a concrete example, one can imagine exciting silicon with a short laser pulse with photon energies greater than the band gap. In this case, the electrons will quickly relax to the conduction band minimum within 10s of femtoseconds, yielding an electronic density that is different from the equilibrium density. It will then take some nanoseconds before the electrons from the conduction band edge recombine with the holes at the valence band maximum. Nuclei, on the other hand, are only capable of moving on the 100s of femtoseconds timescale. Thus, they end up feeling a different electrostatic environment after the initial change in the electronic density which, at least in the case of silicon, appears almost instantaneously and lasts for nanoseconds.
What I am trying to say in words is that the driving force due to the redistribution of electronic density is more appropriately modeled as a Heaviside step function rather than a delta function. So we can write down the following equation with a force that has a step function-like time dependence:

where 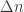

In this case, we can solve this differential equation for 


The solution to this equation gives both sine and cosine terms, but because the light pulse does not change the velocity of the nuclei, we can use the initial condition that 



which this time exhibits a cosine-like oscillation like in the schematic depicted below:

Now that the difference in terms of phase between the two mechanisms is hopefully clear, let’s talk about the symmetry of the modes that can be excited. Because the frequency of light used in these experiments is much higher than the typical phonon frequency, the incident light is not going to be resonant with a phonon. So in this limit (
From a symmetry vantage point, which modes will be excited is determined by the following equation:
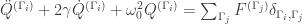
where 

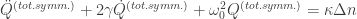
For Raman active modes, the symmetry rules get a little more cumbersome. We can write an expression for the force in terms of the Raman polarizability tensor:

where 


Although this post has now gone on way too long, I hope it helps to serve those who are using laser pulses in their own labs get a start on a topic that is rather difficult to bridge in the existing literature. Please feel free to comment below if there’s anything I can make clearer in the post or expand on in a future post.
Summary of key differences between the two generation mechanisms:
Impulsive stimulated Raman scattering (ISRS):
- Observed in both opaque and transparent crystals
- Away from resonance, oscillations are usually observed with a sine-like phase
- Only Raman-active modes are observed
- Light’s electric field is the “driving force” of oscillations
Displacive excitation of coherent phonons (DECP):
- Observed only if material is opaque (at the frequency of the incoming light)
- Oscillations are observed with cosine-like phase
- Only totally symmetric modes can be excited
- Change in electronic density, and thus a new electrostatic environment, is the “driving force” of oscillations
