Ferromagnetism is a state of matter whose existence has been known for millennia (Fe3O4 supposedly was the first known permanent magnet, but is actually a ferrimagnet). Coincidentally, it was also the material that took center stage in Expt 8 in this series of posts. Antiferromagnetism is, in some sense, “hidden order”. Although predicted in 1932 by Louis Neel, it was not apparent how to convincingly demonstrate that spins could align antiparallel to one another when the magnetic moments perfectly cancel. There is no net magnetization to speak of or measure!
Suggestions of a such a state were hinted at through the observation of a phase transition in magnetic susceptibility measurements, but the origin of such a phase transition was unclear. In 1949, Clifford Shull and J. Stuart Smart definitively demonstrated that antiferromagnetism was present in MnO. In their experiment, which used the new technique of neutron diffraction, magnetic peaks suddenly showed up below 

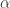 -quartz and
-quartz and  -quartz at 573C was associated with a soft phonon using (you guessed it!) Raman spectroscopy. However, it is important to note that the
-quartz at 573C was associated with a soft phonon using (you guessed it!) Raman spectroscopy. However, it is important to note that the  transition in quartz is a discontinuous phase transition. So while the phonon does soften considerably, it does not actually reach zero frequency before the structural transition takes place.
transition in quartz is a discontinuous phase transition. So while the phonon does soften considerably, it does not actually reach zero frequency before the structural transition takes place.

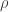 ) vs 1/T in Fe3O4 with different phase fractions of Fe2O3. When the concentration of Fe2O3 is high, the resistivity no longer exhibits a discontinuity.
) vs 1/T in Fe3O4 with different phase fractions of Fe2O3. When the concentration of Fe2O3 is high, the resistivity no longer exhibits a discontinuity. 

